Vorwort Chris: Liebe Leser, heute kommt ein Artikel, der durchaus auch in einem wissenschaftlichen Journal erscheinen können. Daher: Nicht den Mut verlieren und versuchen, wesentliche Aspekte zu verstehen und für das tägliche Leben abzuleiten. Viel Spaß mit dem Artikel unseres Gastautors Tim.
Beim Low-T3-Syndrom führt kein Weg am Gehirn vorbei.
Der aktuelle Artikel kann nicht mit vielen praktischen Tips aufwarten. Dafür liefert er viele historische, mechanistische und hypothetische Ausführungen rund um die Rolle des Gehirns für die ganzkörperliche Stoffwechselgesundheit.
In erster Linie wird es darum gehen, was im Gehirn geschieht, wenn man weit weniger Makronährstoffe (und somit Kalorien) zu sich nimmt als man tatsächlich benötigt.
Hallo Low-T3-Syndrom!
Das alles kann man allerdings auch gerne auf einen Mikronährstoffmangel übertragen. Denn was bringen uns Kohlenhydrate oder Fette, wenn unsere biochemische Maschinerie diese nicht effizient in ATP umsetzen kann?
Während sich manche Leute darüber streiten, ob denn nun zu viele Kohlenhydrate oder zu viele Fette “schuld” an den vielen Gesundheitsproblemen sind, behaupte ich:
Grund ist möglicherweise nicht einmal ein Zuviel an Kalorien.
Ich persönlich denke, es hapert häufig an der Mikronährstoffdichte unserer Nahrung.
Man sollte sich immer ins Bewusstsein rufen, dass trotz Überschuss an “leeren” Kalorien deine Zellen Hunger leiden können. Das Gehirn besteht natürlich auch aus Zellen und diese haben zum einen einen besonders hohen Energiebedarf, zum anderen reagieren sie sehr sensibel auf periphere Hungersignale. Und das kann äußersts gravierende Folgen haben.
Info
Ob Low-T3-Syndrom oder Adipositas: Würde man die biochemische Maschinerie ausreichend “ölen” – keine High-Fat Empfehlung an der Stelle – würden viele Probleme eventuell nicht oder weniger signifikant auftreten.
Bezüglich Adipositas gilt zusätzlich, dass das Stillen der benötigten Mikronährstoffe durch vorzugsweise natürliche Lebensmittel meistens auch einen Kalorienexzess schwer möglich macht.
Außerdem setzt man sich somit automatisch mit der Nahrung intensiver auseinander und zelebriert diese für biochemische Auswirkungen, Geschmack und des Essens wegen (soziokulturelle Faktoren).
Was bisher geschah …
Die vorrausgegangenen Artikel III und IV behandelten, wie in klinisch “gesunden” Menschen eine suboptimale Schilddrüsenfunktion und/oder eine eingeschränkte T3-Wirkung entstehen kann.
Durch Ernährung und Lebensstil können sich diese diffusen Probleme allerdings manifestieren und (i) in einer tatsächlichen Schilddrüsenunterfunktion resultieren oder (ii) im Zuge des Low-T3-Syndroms metabolische und hormonelle Entgleisungen mit sich bringen. Im schlechtesten Fall dauert es einige Zeit um dies wieder auszugleichen und man kämpft mit einer instabilen Psyche, Konzentrationproblemen, einer geringer körperlichen Leistungsfähigkeit, einem schwachen Immunsystem und hoher Infektanfälligkeit, einer reduzierten Libido und Fruchtbarkeit (z.B. Amenorrhoe bei vielen Leistungssportlerinnen) et cetera.
Im abschließenden Artikel will ich nun die Rolle des Gehirns diskutieren und was in Überambitionierten während kalorischer Selbstgeißelung passiert.
Da ich eigentlich Neurobiologie studiert habe, ist das so zu sagen mein “Metier”. Für die Allgemeinheit sind wohl Hormone und die generelle Physiologie deutlich eingängiger. Trotz allem stellt das Gehirn einen wichtigen – wenn nicht sogar DEN wichtigsten – Faktor in der “Gesundheitsgleichung” dar. Das Gehirn vermittelt nicht nur den Feedback der Schilddrüsenachse sondern garantiert langfristig über vielerlei Wege eine gesunde Körperkomposition.
Deshalb und weil das Low-T3-Syndrom im Gehirn signifikante Spuren (oder Narben?!) zu hinterlassen scheint, möchte ich die Reihe mit einem Neuro-Artikel abschließen.
Dieser Artikel auf einen Blick
- Versuchstiere sind notwendig für Analysen des Gehirns.
- Das Gehirn reguliert die Energie Homöostase auf vielerlei Wegen, z.B. über die Schilddrüsenachse.
- Die Konzentration von Leptin (“Sättigungshormon”) korreliert positiv mit der Körperfettmasse und steigert die Schilddrüsenfunktion; Leptin beeinflust im Gehirn die Produktion von TRH (Thyreotropin) direkt und indirekt.
- Ein niedriger Körperfettanteil, starkes Kalorien-(/Kohlenhydrat) Defizit sowie langes Fasten reduziert Leptin, wodurch sekundär der Set-point der Schilddrüsenachse nach unten angepasst wird.
- Cortisol steigt als “Stresshormon” in diesem Kontext an und interferiert mit der TRH-Produktion und der peripheren Schilddrüsenhormonwirkung.
- Kurzer Nahrungsentzug in Nagern ist ein Modell für das Low-T3-Syndrom.
- T3 Spiegel ist im Gehirnbereich “Hypothalamus” während des Fastens lokal erhöht und hemmt die TRH Produktion (und hat weitere Effekte wie stimulierte Nahrugsaufnahme).
- Lokaler Anstieg beruht auf verstärktem Transport von T4 über Blut-Hirnschranke (mehr MCT8 Transporter) und verstärkte Konversion zu T3 (hypothalamische Deiodinase Typ II wird stimuliert).
- Sogenannte AgRP-Neurone sind prominente “Hunger-Zellen” im Hypothalamus; durch lokales T3 wird deren Bioenergetik verändert.
- Mehr Mitochondrien und Wärmebildung durch Entkoppler “UCP2” in diesen Hunger-Zellen.
- Erhöhte ATP-Produktion und Thermogenese in den zugehörigen Nervenfasern zwint Nager zu einem kompensatorischen Überfressen bei Zugang zu Nahrung (“rebound feeding”).
- Hypothese:
Beeinflusst die Temperatur im Hypothalamus die Signalübertragung negativ mit Hinblick auf die Energie Homöostase (thermale Synapsen)?
Abhängig von Ratten und Mäusen:
Was es bei translationaler Erforschung der Schilddrüse zu beachten gilt
Es liegt in der Natur der Sache, dass Humanstudien selten zu einem detailierten Verständnis der
Mechanismen beitragen können. Insbesondere Untersuchungen der Gehirnkomponente sind beim Menschen technisch limitiert. Auch wenn Tierversuchsgegner dies gerne ausblenden ist die Forschung gerade in diesem Bereich gezwungen auf Tiermodelle auszuweichen. Natürlich sind Mäuse und Ratten keine “kleine Menschen”. Der offensichtlichste Unterschied ist die kleine Körpergröße und die höhere Stoffwechselrate. Auf Grund Letzteren muss die Schilddrüsenfunktion von Nagern zwangsläufig deutlich flexibler reagieren können. Wäre nicht eine rapide Hemmung der Schilddrüsenaktivität und damit des Grundumsatzes gegeben, könnten für Nager schon kurze Perioden an Nahrungsknappheit tödlich enden (Flier et al., 2000; Boelen et al., 2008).
Kurzer Nahrungsentzug genügt in Mäusen um T3 stark abfallen zu lassen (Blake et al., 1991; Ahima et al., 1996). Theoretisch würde durch den negativen Feedback die Produktion von TRH und TSH kompensatorisch gesteigert werden um die Schilddrüse wieder anzutreiben.
Dieser Feedback ist aber bei Kalorienrestriktion überschrieben.
In den letzten zwei bis drei Jahrzehnten wurden durch Tiermodelle faszinierende Mechanismen entdeckt, welches dieses Paradoxon zu erklären versuchen. Bevor wir dies behandeln, hier zuerst noch einmal die Veranschaulichung der Situation:
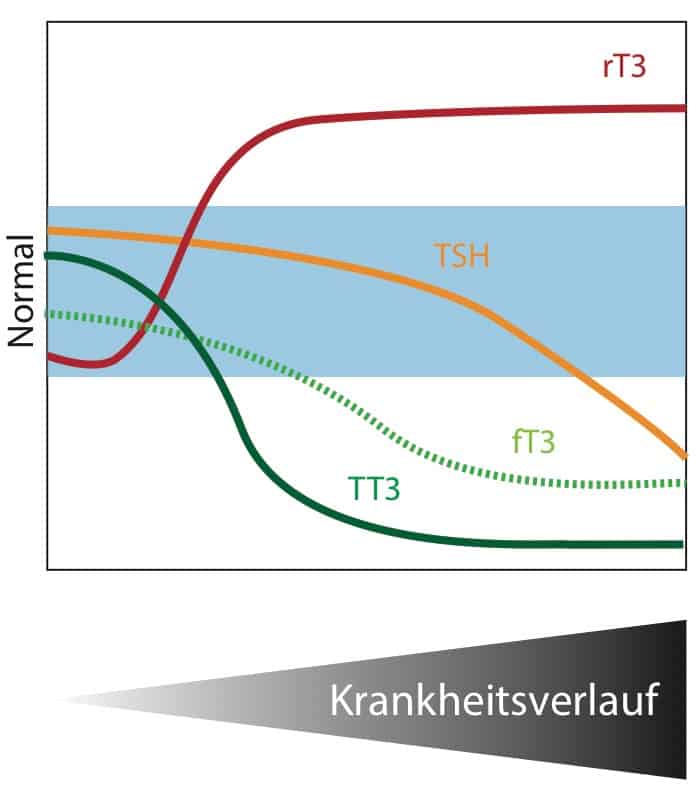
An dieser Stelle sei betont, dass trotz der reduzierten Spiegel der Schilddrüsenhormone (TT3 und fT3) der TSH-Wert nicht kompensatorisch ansteigt beziehungsweise sogar mit zunehmender Schwere der Erkrankung abfallen kann (äquivalent zu Erkrankung: Radikal-Diät, Entzündung,…).
Hormonelle Verschiebungen durch Kaloriendefizit und deren Wahrnehmung durch das Gehirn
Zu Beginn möchte ich die Aufmerksamkeit auf eben dieses Phänomen lenken, warum der Körper trotz reduzierter Schilddrüsenhormonspiegel nicht mit einer kompensatorischen TRH und TSH Freisetzung reagiert. Das ist nämlich auch beim Menschen zu beobachten. Hier bedarf es aber einer etwas längeren Fastendauer um eine Abnahme des TSH und der peripheren Schilddrüsenhormone beobachten zu können (Burman et al, 1980; O’Brian et al., 1980; Spencer et al., 1983).
Info
Diese Thematik – sowohl im Mensch als auch im Nager – wird meiner Meinung nach vortrefflich in einem Review von Boelen, Wiersinga und Fliers diskutiert (Boelen et al., 2008).
Wie wir wissen sitzt die höchste Instanz des thyreotropen Regelkreises im Gehirn, genauer dem Hypothalamus. Dieser ist ein recht gut durchbluteter Teil des Gehirns. An der absoluten Basis des Hypothalamus finden sich Eintrittsstellen für zirkulierende Hormone. Die ansonsten recht hermetische Blut-Hirnschranke weist hier sogenannte fenestrierte Blutgefäße auf. An dieser Schnittstelle können Substanzen zwischen dem Gehirn und dem Körperkreislauf ausgetauscht werden (neurohämale Kontakte). Auch das in Teil II angesprochene Neuropeptid TRH wird hier aus Nervenfasern ins Blut freigesetzt und wirkt in der Folge stimulierend an der Hirnanahangsdrüse, dem Ort der TSH Freisetzung.
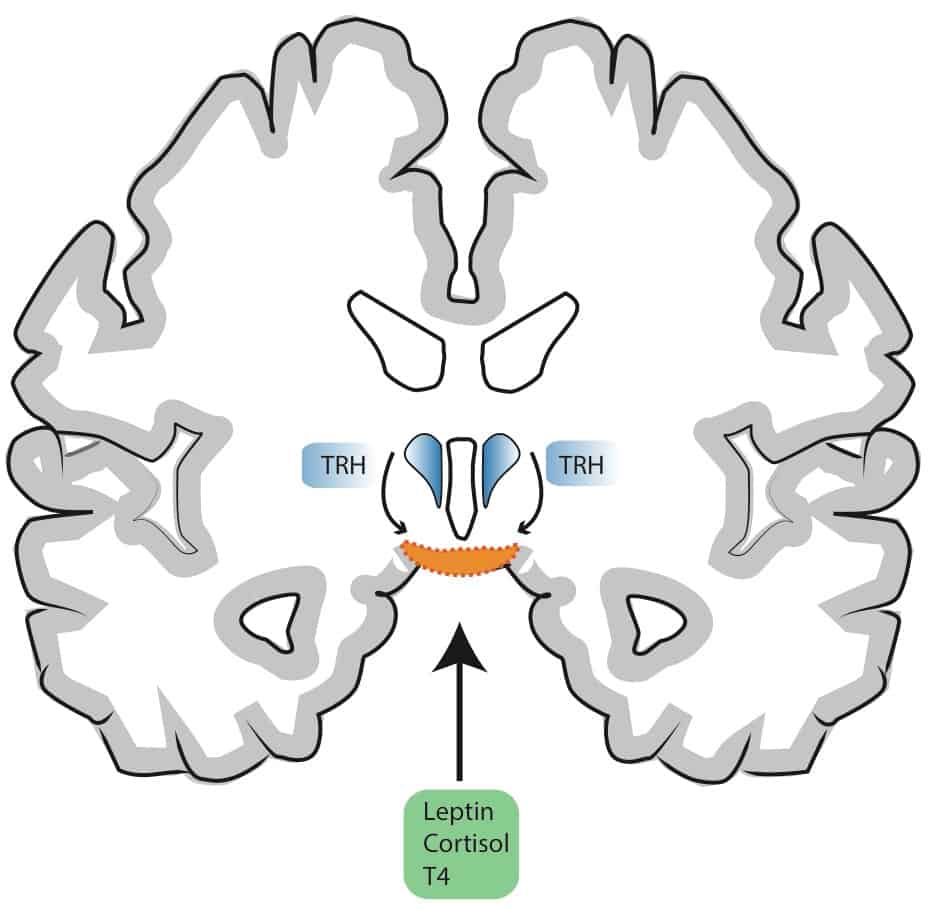
Farblich hervorgehoben sind zwei Strukturen des Hypothalamus:
Der “herzförmige” Nucleus Paraventricularis (blau) findet sich links und rechts des dritten Hirnventrikels und produziert u.a. das Neuropeptid TRH (thyreotropin-releasing hormone).
Die Mediane Eminenz (orange) und anliegende Bereiche an der absoluten Basis des Gehirns dienen als neurohämale Schnittstelle innerhalb der Blut-Hirnschranke (Bennaroch, 2001; Morita & Miyata, 2012; Ciofi, 2011).
Hier werden Substanzen ins Blut ausgeschüttet (z.B. TRH) oder es gelangen zirkulierende Hormone ins Gehirn (z.B. Leptin, Glucocorticoide oder T4).
Leptin – Indikator der Fettmasse und Stoffwechselregulator
Wie doch die Zeit vergeht: Das “Sättigungshormon” Leptin feiert dieses Jahr schon seinen 20. Geburtstag. Aus gegebenem Anlass rekapituliert sein Entdecker, der großartige Jeffrey Friedman, die vergangenen zwei Jahrzehnte, welche dieses Hormon fast so populär werden liesen wie Insulin oder Cortisol (Friedman et al., 2015).
Trotz dieser Popularität möchte ich dennoch einige rudimentäre Grundlagen vermerkt wissen. Leptin ist ein sogenanntes Adipokin (= Cytokin-artig aus dem Fettgewebe stammend). Inzwischen hat man anerkannt, dass das Depotfett nicht nur Speichergewebe ist, sondern zusätzlich ein endokrin-aktives Organ ähnlich einer Hormondrüse. Leptin wird vom (subkutanen) Fettgewebe ins Blut freigesetzt wobei die Leptinmenge positiv mit der Fettmasse korelliert.
Bedeutet banal gesagt:
wenig Depotfett = wenig Leptin = mehr Hunger
viel Depotfett= viel Leptin = weniger Hunger
Somit fungiert dieses Hormon vor allem als Langzeit-Energiesensor. Wie wir heute wissen spielt Leptin eine zentrale Rolle in der Energie Homöostase. Diese entspricht im besten Fall der groben Aufrechterhaltung eines gewissen Körpergewichts beziehungsweise Körperfettanteils. Leptin spielt hierbei das wichtige Feedback-Signal, welches den “Füllstand” der Fettzellen an das Gehirn übermittelt.
Info
Die Adipositas-Forschung wurde von der Entdeckung des Leptins signifikant und maßgeblich geprägt. Das konzeptuelle Verständnis der Fettleibigkeit beruht zu einem Großteil auf Leptin und der Hypothese der gestörten Lipostase, ein Kunstwort für “Körperfett-Balance” (Kennedy, 1953).
Leptins Beziehung zur Schilddrüsenachse:
Die dynamische Veränderungen der Leptinkonzentration ist aber nicht immer direkt proportional zu den langsamen Schwankungen der Körperfettmasse.
Leptin reagiert sehr sensibel auf akute Veränderungen der Ernährungssituation (Weigle et al., 1997; Sinha & Caro, 1998; Chan et al., 2008). Eine dreitägige Fastenphase wird zwar deine Fettspeicher nicht sonderlich reduzieren, wohl aber deine Leptinspiegel drastisch abfallen lassen.
| Explizite Beispiele Sieben schlanke Frauen fasteten für drei komplette Tage. Obwohl sie nur 1,7± 0,6 kg an Körpergewicht verloren, fiel ihr Leptinspiegel in dieser Zeit um 62% (von 8,5± 4.5 auf 2,4± 0,5 ng/ml) (Weigle et al., 1997). Laut anderen Quellen genügen aber auch schon 24 Stunden Fasten um Leptin bis zu 60-70% abzusenken (Sinha & Caro, 1998). Die sieben Frauen erreichten aber innerhalb von 12 Stunden mit isokalorischer Nahrungszufuhr wieder ihre basalen Leptinspiegel (Weigle et al., 1997). Es ist davon auszugehen, dass kurzzeitiges Überessen das zirkulierende Leptin auf 150% der basalen Spiegel anheben kann. Eine weitere Studie – ebenfalls in schlanken Frauen – lässt vermuten, dass vor allem Kohlenhydrate für diesen Leptinanstieg und gesteigerten Energieverbrauch (7%) verantwortlich sind; Fett hingegen bewirkte keine Veränderungen (Dirlewanger et al., 2000). |
Der Abfall der Leptinkonzentration wird im Hypothalamus wahrgenommen (Chan et al., 2008). Ganz besonders die TRH-Nervenzellen reagieren sehr sensibel, da sie die voll-funktionsfähige Form des Leptinrezeptors (ObRb) expremieren (Mercer et al., 1996). Die Aktivierung dieser Leptinrezeptoren wird als essentiell für eine maximale TRH-Produktion angesehen (Guo et al., 2004, Huo et al., 2004). Auch in vivo wurden entsprechende Effekte von Leptin auf die TRH-Produktion unzählige Male dargestellt (Legradi et al., 1997; Perello et al., 2006; Ghamari-Langroudi et al., 2010).
Fehlt jedoch diese Stimulation durch Leptin, signalisiert das den TRH-Zellen einen Nahrungsmangel und die Notwendigkeit Energie zu konservieren. Die TRH-Produktion kommt bei Leptin-Mangel ins Stocken, die Schilddrüsenachse wird nach unten angepasst. Der neue Set-Point der Schilddrüsenhormone lässt den Stoffwechel langsamer bzw. sparsamer werden.
Info
Alternativ scheint im Menschen eine niedrig dosierte Leptin-Substitution während niederkalorischen Diäten den Abfall der zirklierenden Schilddrüsenhormone und des Grundumsatzes sowie weiterer neuroendokrinologischer Anpassungen verhindern zu können (Rosenbaum et al., 2002; Chan et al., 2003; Chan et al., 2008). Ich muss es bei dieser Leserschaft eigentlich nicht erwähnen, aber: Dies stellt keine Empfehlung dar, sondern nur ein weiteren Hinweis für einen kausalen Zusammenhang!
Leptin wird von den Fettzellen produziert, über die Blut-Hirnschranke transportiert und mittels eines regulierten Mechanismus im Hypothalamus verteilt (Balland et al., 2014).
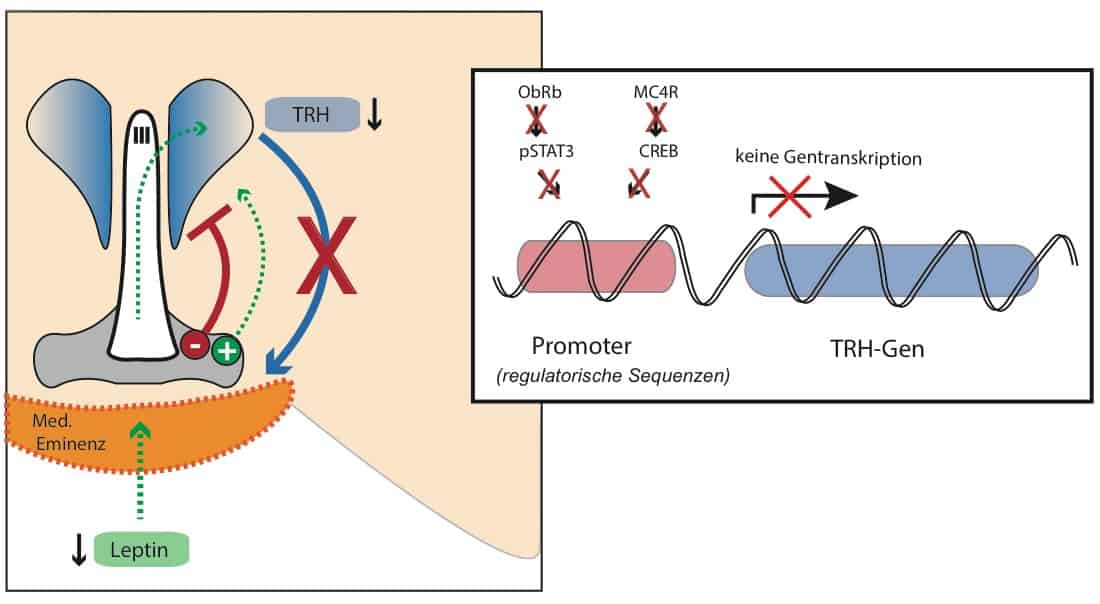
Unter anderem gelangt es über den dritten Hirnventrikel (III) direkt zu den TRH-produzierende Nervenzellen im Ncl. Paraventricularis (blau).Außerdem beeinflusst Leptin an der Basis des dritten Ventrikels die Aktivität von hypothalamischen Schaltkreisen, deren Balance (+/-) unter anderem die TRH-Produktion moduliert. Die Vernetzung der beteiligten Nervenzellen im Hypothalamus ist inzwischen relativ umfassend charakterisiert. Ebenso, wie Leptin auf diese Netzwerke und letzendlich die TRH-Neurone wirkt (Elmquist et al., 2001; Nilni, 2010).
Bei chronischer Unterernährung oder akutem Nahrungsentzug ist der Leptin-Spiegel reduziert (gestrichelte Linie). In der Folge nimmt der stimulierende Input auf die TRH-Produktion ab, wohingegen der inhibitorische Input zunimmt.
| Exkurs für Interessierte: Die zugehörigen Signalwege innerhalb der TRH-Neurone wurden inzwischen entschlüsselt. Leptin wirkt direkt an TRH-Neurone über den langen Leptin-Rezeptor (ObRb) und den JAK2-pSTAT3 Pathway (Huo et al., 2004; Guo et al., 2004)Der indirekte Input über neuronale Schaltkreise wird als Melanocortin-Netzwerk bezeichnet. Er beinhaltet:- stimulierende POMC-Neurone (produzieren α-MSH und CART; grün)- inhibierende AgRP-Neurone (produzieren AgRP, NPY und GABA; rot)Es bestehen eindeutige Hinweise, dass diese Populationen über Nervenfasern funktionale Kontakte mit TRH-Neuronen ausbilden (Toni et al., 1990; Diano et al., 1998; Flier et al., 2000; Fekete & Lechan, 2006; Vella et al., 2011). Die TRH-Neurone produzieren den Melanocortin-Rezeptor (MC4R), welcher in aktiviertem Zustand via CREB die TRH-Produktion steigert. Bei Leptinmangel nimmt der stimulierende Input durch α-MSH ab und der inhibierende AgRP-Input zu. |
Stress – Cortisol interferiert mit der Schilddrüse
Der nächste wichtige Faktor wären Glucocorticoide aus der Nebennierenrinde, welche beim Fasten bzw. Low-Carb Diäten häufig durch die Decke gehen (van Haasteren et al., 1995). Der Mensch bildet in erster Linie das bekannte Cortisol, Nagetiere hauptäschlich Corticosteron. Unabhängig davon zielen beide Stresshormone (unter anderem) darauf ab, Energie durch Abbau von körpereigenem Gewebe bereitzustellen. Man rekrutiert also die Notreserven in Form von Depotfett, Glykogen und körpereigenen Proteinen.
Zusätzlich wirken Glucocorticoide im Hypothalamus als Signalmoleküle. Hier beeinflussen sie wieder einmal die TRH-Nervenzellen. Zu viel Cortisol scheint die Produktion von TRH zu hemmen und senkt die Schilddrüsenaktivität somit schon direkt im Gehirn ab. Auch in der Peripherie scheint Cortisol die Schilddrüsenhormonwirkung auf vielen Ebenen zu antagonisieren.
Bekanntermaßen ist Stress, sogar in Form von Prüfungsangst, destruktiv für die Schilddrüse (Johansson et al. 1987).
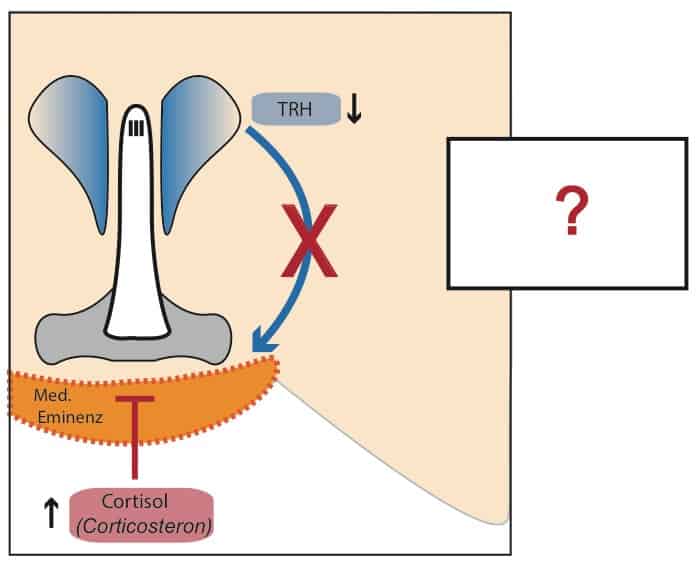
Glucocorticoide wie Cotisol steigen bei Stress und Nahurngsmangel stark an (sofern Nebennierenrinde funktionsfähig ist). Diese Stresshormone interferieren mit den Schilddrüsenhormonen und wirken durch bisher noch nicht ausreichend charakterisierte Mechanismen im Gehirn.
Schilddrüsenhormone im Körper und Gehirn – eine gegensätzliche Regulation überschreibt den Feedback
Das Schilddrüsenhormon T3 wirkt im Gehirn. In Teil II habe ich in einer Infobox schon ein paar Sätze dazu verloren. Vermutlich zu wenig für diesen komplexen Zusammenhang, der mir vor drei Jahren zu Beginn selbst nicht in den Kopf gehen wollte:
Mehr T3 im Hypothalamus wenn man hungert
Ein wesentlicher Grund für die unterdrückte TRH und TSH Freisetzung bei Nahrungsmangel ist der lokal-begrenzte Anstieg von T3 im Hypothalamus. Aber wie kommt es dazu?
Zum einen scheint der Aufnahme von T4 in das Gehirn im Fastenzustand über den Transporter MCT8 selektiv gesteigert zu werden (Copolla et al., 2008). Noch bedeutender ist aber wohl die lokale Aktivierung der Deiodinase Typ II (Ribeiro et al., 2014; Bianco & Kim, 2006; Lechan & Fekete, 2005)
| Zur Erinnerung: Deiodinase Typ II bildet aus T4 das bioaktive T3 (5′-Deiodierung). |
Es ist aktuell nicht genau geklärt, wie genau ein Kaloriendefizit die Deiodinase Typ II selektiv im Hypothalamus zu erhöhen vermag, während eher das Gegenteilige im restlichen Körper zu beobachten ist (s. Teil III; viele Unklarheiten). Glucocorticoide sind eventuell bei dieser lokal-begrenzten Enzymaktiverierung beteiligt (Copolla et al., 2005).
Fakt ist, dass der Körper überlistet wird. Das erhöhte T3 sorgt für eine Unterdrückung der TRH Produktion über den negativen Feedback. Im Normalfall garantiert diese Rückkopplung konstante Hormonspiegel im Serum. Bei Energiemangel spielen MCT8 sowie die Deiodinase Typ II allerdings Lügenbarone um den Feedback zu überschreiben und Energie zu sparen.
Lokal erhöhtes T3 reduziert übrigens nicht nur die TRH-Synthese sondern steigert zusätzlich die Produktion eines TRH-spaltenden Enzyms (Sãnchez et al., 2009).
| Infobox Ein lokaler T3 Anstieg wird übrigens ebenso erreicht, wenn sich im Körper akute oder chronische Entzündungen finden. Eine gestörte Barrierefunktion im Magen-Darm-Trakt steigert die Permeabilität für Bruchstücke von Bakterienzellwänden (Lipopolysaccharide). Diese induzieren eine starke Aktivierung des Immunsystems. Als Schutzmechanismus steigert das Gehirn die lokale Umwandlung von T4 in T3 um die Schilddrüsenachse zu unterdrücken und um Energie zu konservieren (Warner & Beckett, 2009).Auch die Temperatur und die Jahreszeit scheinen eine Rolle zu spielen und verändern die Ökonomie der Schilddrüsenhormone im Hypothalamus. Das lassen Beobachtungen in einigen speziellen Tiermodellen vermuten (z.B. Dschungarischer Hamster). Es wurde zum Beispiel gezeigt, dass die Tageslänge Einfluss auf den Transport und die Deiodierung von Schilddrüsenhormone im Gehirn nimmt. Reguliert werden diese saisonale Schwankungen der hypothalamischen T3 Spiegel durch den T4-Transporter OATP1c1, die Typ II Deiodinase, den Rezeptor GPR50 und viele weitere Faktoren (Bolborea & Dale, 2013; Ebling, 2015).In den entsprechenden Tieren führt dies im Sommer zu mehr T3 im Gehirn, weniger Schilddrüsenhormone in der Peripherie und somit in einem höheren Körperfettanteil . Bevor es in den harschen Winter geht, sind die Tiere folglich mit einem adäquaten Fettdepot ausgestattet. Im Winter weisen sie weniger T3 im Gehirn (und mehr in der Peripherie) auf, um katabole und vor allem thermogene Reaktionen zu induzieren. Die Übertragbarkeit auf den Menschen ist an dieser Stelle allerdings schwierig. |
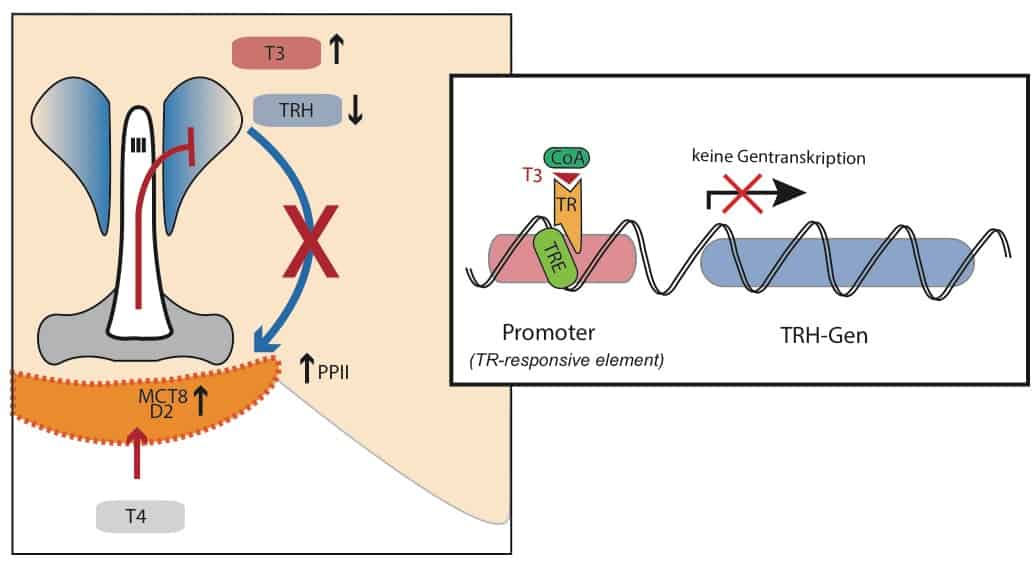
Schilddrüsenhormone werden in das Gehirn transportiert um über einen negativen Feedback den peripheren Ist-Wert konstant zu halten. Trotz abfallenden peripheren Konzentrationen wird im Gehirn bei Nahrungsmangel verstärkt T3 gebildet. Durch diese lokale Wirkung wird die weitere Bildung von TRH unterdrückt. Vermittelt wird diese Hemmung der Achse über aufreguliertes MCT8-Transporter und die Deiodinase Typ II (Bolborea & Dale, 2013). Lokales T3 induziert zusätzlich die Aktivität von Pyroglutamyl Peptidase II (PPII) – ein Enzym, welches ausgeschüttetes TRH inaktiviert (Sãnchez et al., 2009).
Zusammengefasst wird die Schilddrüsenachse in kritischen Zuständen wie Kalorienmangel (oder Entzündungen) nach unten angepasst.
Von zentraler Bedeutung ist (i) ein Leptin-Mangel, (ii) ein Cortisol-Übschuss und (iii) ein lokaler T3 Überschuss.
Wenn das Gehirn heiß läuft – Das Konzept der Thermalen Synapse
An dieser Stelle möchte ich eine äußerst spannende Hypothese beschreiben. Sie stammt aus der Feder eines renommierten Wissenschaftlers der Yale Universität und wird inzwischen auch empirisch gestützt.
“Wärmebildung wird im Gehirn z.B. durch lokales T3 vermittelt. Trägt diese hypothalamische Wärmebildung zur Entstehung von Adipositas bei?”(Horvath et al.,2014).
Schon früh stellte man ausgeprägte Temperaturunterschiede im Gehirn fest. Diese hängen sowohl von der Hirnaktivität als auch schlicht vom betrachteten Gehirnareal ab (Wang et al., 2014). Die Hirnrinde ist grundsätzlich kühler als subcortikale Bereiche (=im Gehirn tiefer-liegend). Interessanterweise ist dieser Gradient nicht linear; vielmehr stellt man einen abrupten Anstieg der Temperatur fest. Die Frage ist: Warum?
Dieser Anstieg der Wärmeproduktion fällt mit dem Auftreten eines gewissen Proteins zusammen, dem uncoupling protein 2 (UCP2) (Horvath et al., 1990).
Schon länger kennt man ein ähnliches Protein aus dem braunen Fett. Dort entkoppelt das besser erforschte UCP1 die mitochondriale Atmungskette und generiert somit Wärme.
Sein Bruder UCP2 findet sich in verschiedenen Geweben, darunter wie erwähnt das Gehirn.
Das Maß der Entkopplung (=Wärmebildung) im Hypothalamus ist zum einen kontext-abhängig und zum anderen evenuell individuell.
Je nach eigener Prädisposition und Lebensstil könnte weitreichende Implikationen haben. Ein wärmerer Hypothalamus begünstigt nämlich die Nahrungszufuhr und verlangsamt den Stoffwechsel. Salopp gesagt.
Evidenz? Bitte sehr!
Seit Teil I wissen wir, dass die Wärmebildung im braunen Fettgewebe sehr ausgeprägt durch Schilddrüsenhormone beeinflusst wird (T3 erhöht UCP1 & Sensitivität brauner Fettzellen gegenüber Catecholamine). Ähnlich wie im braunen Fett wird auch im Gehirn die Wärmebildung durch T3 begünstigt. Verglichen mit dem braunen Fett ist hier die erhöhte Produktion von UCP2 in gewissen Nervenzellen nicht sonderlich wünschenswert (Copolla et al., 2007).
Info
Man sollte erwähnen, dass nicht alle Nervenzellen im Hypothalamus gleich sensibel auf lokale T3 Anstiege reagieren. Viele Zellen sind in der Lage die intrazellulären (!!!) T3 Spiegel stabil zu halten indem sie das Enzym “Deiodinase Typ III” produzieren, welches zu viel T3 inaktivieren kann (Kalló et al., 2014). Vereinzelte Zellen scheinen dieses Enzym allerdings nicht zu besitzen. Somit stellt für sie ein T3 Anstieg ein starkes Signal dar. Zum Beispiel ein Signal um UCP2 zu produzieren.
“Hunger-Nervenzellen” werden aufgeheizt
Diese UCP2 Produktion geschieht besonders in einem gewissen Zelltypus, den sogenannten AgRP/NPY-Neuronen. Sie stellen diejenigen Nervenzellen dar, welche dich regelrecht zum Essen “zwingen”. Dies gelingt ihnen durch die Freisetzung orexigener (=Hunger-auslösender) Botenstoffe im Gehirn, welche ebenfalls die Namensgeber dieser Zellen sind (AgRP/NPY: von Agouti-related Peptide und Neuropeptide Y).
Gerade eben erst haben wir ein Modell kennengelernt, durch welches man elegant einen körpereigenen T3 Anstieg im Hypothalamus verursachen kann: Nahrungsentzug!
Es wurde eindrucksvoll gezeigt, dass ein T3 Anstieg im Fastenzustand eine vermehrte Bildung von Mitochondrien in AgRP/NPY-Nervenzellen induziert, die zusätzlich via UCP2 stark entkoppelt sind (Copolla et al., 2007). Somit weisen AgRP/NPY-Neurone gegen Ende des Fastens eine veränderte Bioenergetik auf. Die Autoren der Studie sammelten Hinweise, dass die lokal-produzierte Wärme stimulierend auf die neuronale Aktivität der “Hunger-Zellen” wirkt.
Info
Die Kommunikation im Gehirn wie z.B. die synaptische Transmission ist ein energieaufwändiger Prozess (Mergenthaler et al., 2013). Mitochondrien finden sich folglich besonders in den Nervenfasern und Prä-Synapsen um die Signalübertragung am Laufen zu halten.Um das in Relation setzen zu können:
Die Freisetzung eines einzigen synaptischen Vesikels erfordert die Hydrolyse von 1.64 x105 Moleküle ATP (Harris et al., 2012)
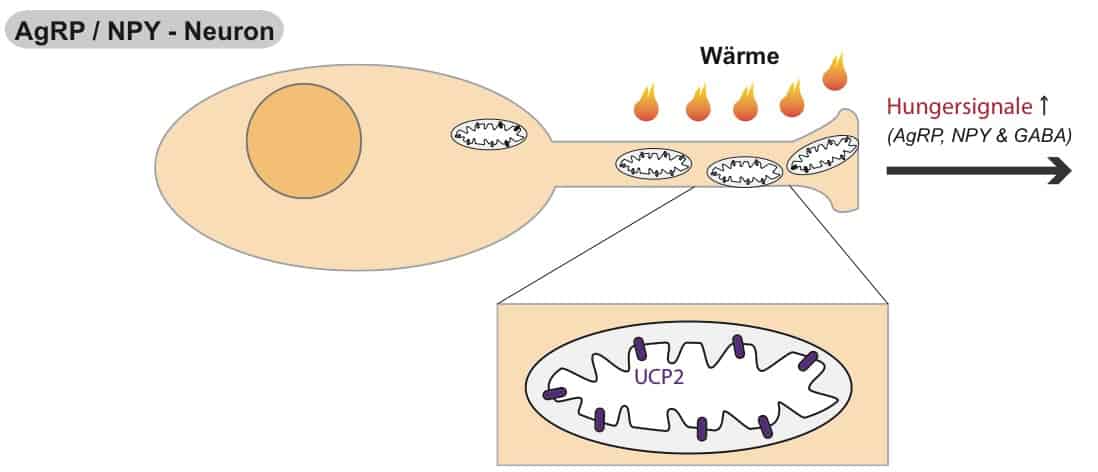
Zusätzlich konnten zwei Folgestudien einen kausalen Zusammenhang zwischen UCP2 und einem reduzierten oxidativen Stress darstellen. Neben der mitochondrialen Biogenese und Wärmebildung scheint das Abfangen von reaktiven Sauerstoffspezies Aktivität der AgRP/NPY-”Hungerzellen” stimulieren. Wir wissen aus Teil I, dass eine Entkopplung der mitochondrialen Atmungskette (z.B. durch UCP2) die Bildung von reaktiven Sauerstoffspezies (ROS) reduziert. In diesen Studien wurde gezeigt, dass weniger ROS im Hypothalamus zu einer gesteigerten Nahrungsaufnahme führt (Andrews et al., 2009).
Nicht sonderlich überraschend werden sich die gefasteten Ratten kompensatorisch überfressen sobald ihnen wieder Zugang zum Futter gewährt wird. Die bioenergetischen Veränderungen in AgRP/NPY-Neurone bilden eine der neurophysiologischen Grundlagen dieses Phänomens. Ihre persistierende Aktivität begünstigt die Nahrungsaufnahme – selbst wenn die Futtermenge im Normalfall schon längst die Sättigung hätte induzieren sollen.
Interssanterweise trifft auch das Gegenteil zu – vermehrter oxidativer Stress im Hypothalamus aktiviert Sättigungssignale über peroxisomale Mechanismen in Nachbarzellen (Diano et al., 2011).
ROS sind also nicht pauschal schlecht, sondern in gewissem Umfang auch bedeutende Signalstoffe.
Wird durch Fasten das menschliche Gehirn ähnlich negativ aufgeheizt?
Ich denke, dass zu häufiges und/oder zu extremes Fasten durchaus ähnlich negative Mechanismen im Gehirn induzieren könnte. AgRP/NPY Nervenzellen haben vielerlei Aufgaben wie z.B. (i) Vermittlung von Hunger, (ii) Hemmung der Schilddrüsenachse oder (iii) Steigerung der endogene Glucose-Produktion durch die Leber.
Sollte man deren Aktivität ungünstig steigern könnten sich hypothetische Probleme wie Essstörungen (binge eating), das Low-T3-Syndrom oder eine verschlechterte Blutzuckerkontrolle ergeben. Ab und an eine Fastenphase einzubauen hat sicherlich gesundheitliche Vorteile (AMPK, Sirtuine, PGC1α,…). Trotzdem gilt wie überall: Maß halten.
Auch ÜBergewicht und Adipositas könnte den Hypothalamus aufheizen. Hier ist dann eventuell nicht der Fasten-induzierte T3 Anstieg der Übeltäter, sondern die Abundanz an Energieträger. Deren unvollständige Oxidation kann zu oxidativem Stress führen, welche Zellen im Gehirn durch UCP2-Entkopplung abzumildern versuchen.
Info
Man könnte weiters spekulieren, wie genetisch-determinierte Faktoren und Umweltfaktoren zusammenwirken um die Verschaltung und Temperatur im Hypothalamus zu beeinflussen. Das könnte (hypothetisch!) eine individuelle Prädisposition für Adipositas oder auch das Low-T3-Syndrom erklären.
Referenzen
Ahima, Rexford S. et al. ‚Role Of Leptin In The Neuroendocrine Response To Fasting‘. Nature 382.6588 (1996): 250-252. Web.
Andrews, Zane B. et al. ‚UCP2 Mediates Ghrelin’s Action On NPY/Agrp Neurons By Lowering Free Radicals‘. Nature 459.7247 (2009): 736-736. Web.
Balland, Eglantine et al. ‚Hypothalamic Tanycytes Are An ERK-Gated Conduit For Leptin Into The Brain‘. Cell Metabolism 19.2 (2014): 293-301. Web.
Benarroch, E. E. ‚Circumventricular Organs: Receptive And Homeostatic Functions And Clinical Implications‘. Neurology 77.12 (2011): 1198-1204. Web.
Bianco, A. C., and B. W. Kim. ‚Deiodinases: Implications Of The Local Control Of Thyroid Hormone Action‘. Journal of Clinical Investigation 116.10 (2006): 2571-2579. Web.
Blake, N. G. et al. ‚Inhibition Of Hypothalamic Thyrotropin-Releasing Hormone Messenger Ribonucleic Acid During Food Deprivation‘. Endocrinology 129.5 (1991): 2714-2718. Web.
Boelen, Anita, Wilmar Maarten Wiersinga, and Eric Fliers. ‚Fasting-Induced Changes In The Hypothalamus-Pituitary-Thyroid Axis‘. Thyroid 18.2 (2008): 123-129. Web.
Bolborea, Matei, and Nicholas Dale. ‚Hypothalamic Tanycytes: Potential Roles In The Control Of Feeding And Energy Balance‘. Trends in Neurosciences 36.2 (2013): 91-100. Web.
Burman, Kenneth D. et al. ‚Nature Of Suppressed TSH Secretion During Undernutrition: Effect Of Fasting And Refeeding On TSH Responses To Prolonged TRH Infusions‘. Metabolism 29.1 (1980): 46-52. Web.
Chan, J. L. et al. ‚Differential Regulation Of Metabolic, Neuroendocrine, And Immune Function By Leptin In Humans‘. Proceedings of the National Academy of Sciences 103.22 (2006): 8481-8486. Web.
Chan, Jean L, Shekman L Wong, and Christos S Mantzoros. ‚Pharmacokinetics Of Subcutaneous Recombinant Methionyl Human Leptin Administration In Healthy Subjects In The Fed And Fasting States‘. Clinical Pharmacokinetics 47.11 (2008): 753-764. Web.
Chan, Jean L. et al. ‚The Role Of Falling Leptin Levels In The Neuroendocrine And Metabolic Adaptation To Short-Term Starvation In Healthy Men‘. Journal of Clinical Investigation 111.9 (2003): 1409-1421. Web.
Chan, Jean L. et al. ‚The Role Of Falling Leptin Levels In The Neuroendocrine And Metabolic Adaptation To Short-Term Starvation In Healthy Men‘. Journal of Clinical Investigation 111.9 (2003): 1409-1421. Web.
Ciofi, Philippe. ‚The Arcuate Nucleus As A Circumventricular Organ In The Mouse‘. Neuroscience Letters 487.2 (2011): 187-190. Web.
Cone, R D et al. ‚The Arcuate Nucleus As A Conduit For Diverse Signals Relevant To Energy Homeostasis‘. Int J Obes Relat Metab Disord 25 (2001): S63-S67. Web.
Coppola, Anna et al. ‚A Central Thermogenic-Like Mechanism In Feeding Regulation: An Interplay Between Arcuate Nucleus T3 And UCP2‘. Cell Metabolism 5.1 (2007): 21-33. Web.
Coppola, Anna et al. ‚Suppression Of Hypothalamic Deiodinase Type II Activity Blunts TRH mRNA Decline During Fasting‘. FEBS Letters 579.21 (2005): 4654-4658. Web.
Diano, Sabrina et al. ‚Monosynaptic Pathway Between The Arcuate Nucleus Expressing Glial Type II Iodothyronine 5′-Deiodinase mRNA And The Median Eminence-Projective TRH Cells Of The Rat Paraventricular Nucleus‘. Journal of Neuroendocrinology 10.10 (1998): 731-742. Web.
Diano, Sabrina et al. ‚Peroxisome Proliferation-Associated Control Of Reactive Oxygen Species Sets Melanocortin Tone And Feeding In Diet-Induced Obesity‘. Nat Med 17.9 (2011): 1121-1127. Web.
Dirlewanger, M et al. ‚Effects Of Short-Term Carbohydrate Or Fat Overfeeding On Energy Expenditure And Plasma Leptin Concentrations In Healthy Female Subjects‘. Int J Obes Relat Metab Disord 24.11 (2000): 1413-1418. Web.
Ebling, Francis J.P. ‚Hypothalamic Control Of Seasonal Changes In Food Intake And Body Weight‘. Frontiers in Neuroendocrinology 37 (2015): 97-107. Web.
Elmquist, Joel K. ‚Hypothalamic Pathways Underlying The Endocrine, Autonomic, And Behavioral Effects Of Leptin‘. Physiology & Behavior 74.4-5 (2001): 703-708. Web.
Fekete, Csaba, and Ronald M. Lechan. ‚Neuroendocrine Implications For The Association Between Cocaine- And Amphetamine Regulated Transcript (CART) And Hypophysiotropic Thyrotropin-Releasing Hormone (TRH)‘. Peptides 27.8 (2006): 2012-2018. Web.
Flier, Jeffrey S., Mark Harris, and Anthony N. Hollenberg. ‚Leptin, Nutrition, And The Thyroid: The Why, The Wherefore, And The Wiring‘. Journal of Clinical Investigation 105.7 (2000): 859-861. Web.
Friedman, Jeffrey M., and Christos S. Mantzoros. ’20 Years Of Leptin: From The Discovery Of The Leptin Gene To Leptin In Our Therapeutic Armamentarium‘. Metabolism 64.1 (2015): 1-4. Web.
Ghamari-Langroudi, Masoud et al. ‚Regulation Of Thyrotropin-Releasing Hormone-Expressing Neurons In Paraventricular Nucleus Of The Hypothalamus By Signals Of Adiposity‘. Endocrine Reviews 31.6 (2010): 942-942. Web.
Guo, Feifan et al. ‚Leptin Signaling Targets The Thyrotropin-Releasing Hormone Gene Promoter In Vivo‘. Endocrinology 145.5 (2004): 2221-2227. Web.
Harris, Julia J., Renaud Jolivet, and David Attwell. ‚Synaptic Energy Use And Supply‘. Neuron 75.5 (2012): 762-777. Web.
Horvath, Tamas L. et al. ‚Brain Uncoupling Protein 2: Uncoupled Neuronal Mitochondria Predict Thermal Synapses In Homeostatic Centers.‘. J Neurosci 19(23) (1990): 10417-27. Web. 19 May 2015.
Horvath, Tamas L., Nina S. Stachenfeld, and Sabrina Diano. ‚A Temperature Hypothesis Of Hypothalamus-Driven Obesity‘. Yale J Biol Med 87(2) (2014): 149-158. Web. 19 May 2015.
Huo, Lihong et al. ‚Role Of Signal Transducer And Activator Of Transcription 3 In Regulation Of Hypothalamic Trh Gene Expression By Leptin‘. Endocrinology 145.5 (2004): 2516-2523. Web.
Johansson, G et al. ‚Examination Stress Affects Plasma Levels Of TSH And Thyroid Hormones Differently In Females And Males.‘. Psychosomatic Medicine 49.4 (1987): 390-396. Web.
Kalló, Imre et al. ‚A Novel Pathway Regulates Thyroid Hormone Availability In Rat And Human Hypothalamic Neurosecretory Neurons‘. PLoS ONE 7.6 (2012): e37860. Web.
Kennedy, G. C. ‚The Role Of Depot Fat In The Hypothalamic Control Of Food Intake In The Rat‘. Proceedings of the Royal Society B: Biological Sciences 140.901 (1953): 578-592. Web.
Lechan, Ronald M., and Csaba Fekete. ‚Role Of Thyroid Hormone Deiodination In The Hypothalamus‘. Thyroid 15.8 (2005): 883-897. Web.
Legradi, G. ‚Leptin Prevents Fasting-Induced Suppression Of Prothyrotropin-Releasing Hormone Messenger Ribonucleic Acid In Neurons Of The Hypothalamic Paraventricular Nucleus‘. Endocrinology 138.6 (1997): 2569-2576. Web.
Mathieson, Ruth A. et al. ‚The Effect Of Varying Carbohydrate Content Of A Very-Low-Caloric Diet On Resting Metabolic Rate And Thyroid Hormones‘. Metabolism 35.5 (1986): 394-398. Web.
Mercer, Julian G. et al. ‚Localization Of Leptin Receptor Mrna And The Long Form Splice Variant (Ob-Rb) In Mouse Hypothalamus And Adjacent Brain Regions By In Situ Hybridization‘. FEBS Letters 387.2-3 (1996): 113-116. Web.
Mergenthaler, Philipp et al. ‚Sugar For The Brain: The Role Of Glucose In Physiological And Pathological Brain Function‘. Trends in Neurosciences 36.10 (2013): 587-597. Web.
Morita, Shoko, and Seiji Miyata. ‚Different Vascular Permeability Between The Sensory And Secretory Circumventricular Organs Of Adult Mouse Brain‘. Cell Tissue Res 349.2 (2012): 589-603. Web.
Nillni, Eduardo A. ‚Regulation Of The Hypothalamic Thyrotropin Releasing Hormone (TRH) Neuron By Neuronal And Peripheral Inputs‘. Frontiers in Neuroendocrinology 31.2 (2010): 134-156. Web.
O’Brian, John T. et al. ‚Thyroid Hormone Homeostasis In States Of Relative Caloric Deprivation‘. Metabolism 29.8 (1980): 721-727. Web.
Perello, Mario, Ronald C. Stuart, and Eduardo A. Nillni. ‚The Role Of Intracerebroventricular Administration Of Leptin In The Stimulation Of Prothyrotropin Releasing Hormone Neurons In The Hypothalamic Paraventricular Nucleus‘. Endocrinology 147.7 (2006): 3296-3306. Web.
Rosenbaum, M. ‚Low Dose Leptin Administration Reverses Effects Of Sustained Weight-Reduction On Energy Expenditure And Circulating Concentrations Of Thyroid Hormones‘. Journal of Clinical Endocrinology & Metabolism 87.5 (2002): 2391-2391. Web.
Sãnchez, Edith et al. ‚Tanycyte Pyroglutamyl Peptidase II Contributes To Regulation Of The Hypothalamic-Pituitary-Thyroid Axis Through Glial-Axonal Associations In The Median Eminence‘. Endocrinology 150.5 (2009): 2283-2291. Web.
Schurgin, Sunita et al. ‚Endocrine And Metabolic Effects Of Physiologic R-Methuleptin Administration During Acute Caloric Deprivation In Normal-Weight Women‘. The Journal of Clinical Endocrinology & Metabolism 89.11 (2004): 5402-5409. Web.
Spencer, Carole A. et al. ‚Dynamics Of Serum Thyrotropin And Thyroid Hormone Changes In Fasting‘. The Journal of Clinical Endocrinology & Metabolism 56.5 (1983): 883-888. Web.
Toni, Roberto, Ivor M.D. Jackson and Ronald M. Lechan. ‚Neuropeptide-Y-Immunoreactive Innervation Of Thyrotropin-Releasing Hormone-Synthesizing Neurons In The Rat Hypothalamic Paraventricular Nucleus‘. Endocrinology 126.5 (1990): 2444-2453. Web.
van Haasteren, G A C et al. ‚Starvation-Induced Changes In The Hypothalamic Content Of Prothyrotrophin-Releasing Hormone (Pro-TRH) mRNA And The Hypothalamic Release Of Pro-TRH-Derived Peptides: Role Of The Adrenal Gland‘. Journal of Endocrinology 145.1 (1995): 143-153. Web.
Vella, Kristen R. et al. ‚NPY And MC4R Signaling Regulate Thyroid Hormone Levels During Fasting Through Both Central And Peripheral Pathways‘. Cell Metabolism 14.6 (2011): 780-790. Web.
Wang, Huan et al. ‚Brain Temperature And Its Fundamental Properties: A Review For Clinical Neuroscientists‘. Frontiers in Neuroscience 8 (2014): n. pag. Web.
Warner, M. H, and G. J Beckett. ‚Mechanisms Behind The Non-Thyroidal Illness Syndrome: An Update‘. Journal of Endocrinology 205.1 (2009): 1-13. Web.
Weigle, D. S. ‚Effect Of Fasting, Refeeding, And Dietary Fat Restriction On Plasma Leptin Levels‘. Journal of Clinical Endocrinology & Metabolism 82.2 (1997): 561-565. Web.
7 comments On Das Low-T3-Syndrom aus der Neuroperspektive
Daaanke genau das Problem habe ich die ganze Zeit das Binge Eating am Abend… ich folge dem CBL 1.0 programm von john kiefer! Und extreme Mühen während der ulchf phase bis am Abend wenn ich dann nach dem Training esse kann ich nicht mehr aufhören …
habt Ihr irgend ein tip für mich?
Ein anderes Ernährungskonzept vllt….?! ;-)
Oder am besten überhaupt kein Konzept (s. Chris‘ neusten Kommentar)
Ich hab mich mit CBL sowohl theoretisch als auch praktisch befasst und kann sagen, dass es
a) vom wissenschaftlichen Hintergrund sehr wacklig ist und
b) auf Dauer es problematisch sein kann und häufig in Essstörungen resultiert (s. Binge-Eating).
Mag vielleicht einige geben, die gefestigt und reif genug sind sich unter Kontrolle zu haben. Würde aber tippen, dass der Großteil – gerade wenn im kcal Defizit – da regelmäßig Probleme bekommen wird.
Außerdem ist es auch ernährungsphysiologisch mehr als logisch, dass diese Mengen für Probleme sorgen. Gerade die vielen Carbs in Form von Stärke auf einmal. Das wird der Darmflora nie und nimmer gut tun.
Über diesen Aspekt werde ich bald mal einen Artikel schreiben – über Kohlenhydrat-Dichte und wie „zelluläre“, CHO-dichte Lebensmittel im Vergleich zu whole-food Probleme in Bezug der Darmflora machen kann.
Ok,vielen Dank für die Info! Vieles macht zwar Sinn so wie ich finde z.B. Mit den kohlenhydraten nach dem Training. Die Menge finde ich auch fraglich esse einfach bis ich satt bin und achte nicht gross auf zahlrn wenn ich Hunger hab esse ich ansonsten lass ichs sein. Wonit ich Mühe habe ist mit seiner Ansicht der Ernährung beim morgendlichen Training was ist deine Meinung dazu?
Habe das Buch sofort bestellt kann nicht abwarten endlich mal n gesheites Buch auf Deutsch in den Händen zu halten und nich immer alle Infos in Englisch holen zu Müssen!
Sehr interessanter Artikel. Die beschriebenen Szenarien (binge eating, Gewichtsabnahme geht im Winter leichter ) hab ich am eigenen Körper erfahren. Vielen Dank für diesen hervorragenden Artikel ( einen kurzen Satz zur Leptinresistenz hätte ich gut gefunden, würde mich sehr über einen Artikel zum Thema freuen ).
Musste gerade schmunzeln, da ich ursprünglich genau dazu auch einen Exkurs hatte (halbe DinA4 Seite etwa).
Ich wollte aber unbedingt vermeiden, den Artikel „unnötig“ aufzublasen – ist schon umfassend genug ;-)
Natürlich wollte ich Leptin zwar erwähnen, aber nicht den Hauptkriegsschauplatz „Low-T3 Syndrom“ zu stark aus den Augen zu verlieren. Und eine Leptin Resistenz ist in diesem Zusammenhang leider nicht wirklich relevant.
Aber ja…Natürlich ist die Leptinresisistenz ein heißes Thema und wenn man über „mehr Leptin –> weniger Hunger“ schreibt, sollte man das eigtl ausführen.
Ich werde wohl darüber schreiben. Hab den Exkurs noch. Es hat sich in dem Feld inzwischen einiges getan und man kennt spannende Mechanismen. Zusätzlich ist das Problem einer klaren Definition von“klinischer Leptin-Resistenz“ nicht so einfach und darüber liese sich auch einiges schreiben.
Für Insulin ist das klarer definiert und man hat verschiedene Methoden um eine verschlechterte Insulinwirkung und Blutzuckerkontrolle darzustellen (oder eine inadäquate Sekretionsleistung der Beta-Zellen).
Das wäre der oraler Glukose-Toleranztest, HOMA-IR, HbA1c, euglykämische-hyperinsulinämische Clamp’s usw.
Für Leptin ist das schwieriger und ich wäre wie gesagt motiviert, darüber etwas zu schreiben.
In Zukunft will ich aber auch beginnen den ein oder anderen weniger „anspruchsvollen“ Artikel zu schreiben.
Wie Chris sagt ist das ein recht deftiges Niveau. War aber auch der Abschluss der Reihe.
Hallo Bettina,
hier findest du eine recht aktuelle und verständliche Zusammenstellung zu den Ursachen der Leptinresistenz von Stephen Guyenet, die dich interessieren könnte:
https://www.youtube.com/watch?t=2024&v=WMdSHNnRbEs
Vielen Dank für den Artikel! Sehr verständlich erklärt und anschaulich mit Bildmaterial aufgewertet, gefällt mir!