
Heute möchte ich euch eine Theorie vorstellen, über die ich bereits etwas länger nachdenke und die — aus der Sicht der Literatur — definitiv valide ist. Viele Aspekte habe ich vereinzelt bereits angesprochen. Heute möchte ich sie gerne etwas ausführlicher darstellen.
Im Grunde postuliert die Hypothese, dass ein dysfunktionaler L-Carnitin-Stoffwechsel die Weichen für eine langfristige mitochondriale Maladaptation stellt, an dessen Ende die muskuläre Insulinresistenz steht und eine adäquate metabolische Flexibilität verhindert.
Die Hintergründe
Eine klassische metabolische Entgleisung liegt dann vor, wenn Mitochondrien die Substratwahl nicht mehr adäquat anpassen können. Die metabolische Flexibilität. Das Primärsubstrat ist dann die Fettsäure. Das ist kein Problem. Doch diese Mitochondrien reagieren nicht adäquat im Zuge einer Anreicherung von Pyruvat, dem Abbauprodukt aus dem Glukose-Stoffwechsel. Selbst dann, wenn die zelluläre Botschaft kommt, „Achtung, jetzt kommt eine Ladung Kohlenhydrate, die wir oxidieren müssen“, bleibt das Primärsubstrat die Fettsäure.
Bei Gesunden ist das kein Problem. Mitochondrien oxidieren linear zum jeweiligen Substrat-Influx. Wenn also gerade das Kohlenhydrat der dominante Makronährstoff ist, wird ebendieser oxidiert und die Fettsäure-Oxidation nimmt reziprok ab.
Die metabolische Inflexibilität geht — paradoxerweise — sehr häufig einher mit einer defekten mitochondrialen ß-Oxidation. Die Fettverbrennung funktioniert nicht adäquat. Das heißt, Mitochondrien zeigen eine eingeschränkte Fähigkeit zur Oxidation von Fettsäuren und das, obwohl sie eine Präferenz für Fettsäuren zeigen.
Hieraus ergibt sich ein zelluläres Chaos, wobei die ATP-Produktion aus Glukose lahmgelegt ist und die ß-Oxidation trotz einer Fettsäure-Präferenz ebenfalls nicht effektiv funktioniert. Die Folge ist ein latenter Energiemangel trotz massivem Substratüberfluss, dabei entstehen freie Radikale (ROS) und unvollständig oxidierte Fettsäuren reichern sich in den Zellen an. Die wiederum blockieren das Insulin-Signal so, dass nunmehr sehr viel weniger Glukose in die Zellen gelangt.
Diese Sicht zeigt ganz klar auf, dass insulinresistente Individuen zu viele Fettsäuren relativ zur Kapazität oxidieren, diese Oxidation allerdings nicht vollständig ist. Es entsteht ein sogenannter mitochondrial overload.
Zusammengefasst:
- Metabolische Entgleisung geht einher mit metabolischer Inflexibilität
- Das bedeutet, dass die Zelle und das Mitochondrium nicht mehr auf die aktuelle Substratzufuhr reagiert
- Gleichzeitig sehen wir (sehr häufig) eine defekte mitochondriale ß-Oxidation
- Insgesamt ergibt sich eine schlechte energetische Versorgung der Zellen
- (Unvollständig oxidierte) Fettsäuren reichern sich in den Zellen an
- Daraus folgt: Das Insulin-Signal „wirkt“ nicht mehr
Hintergründe zu L-Carnitin
L-Carnitin kann endogen, in der Leber, synthetisiert werden. Die Synthese-Raten sind zwar niedrig, es wird generell aber angenommen, dass sie ausreichen, wenngleich die exogene Zufuhr den endogen synthetisierten Wert bei Weitem übersteigt und der Großteil des L-Carnitins tatsächlich aus der Nahrung stammt.
Für die körpereigene Biosynthese sind vier Enzyme und die Mikronährstoffe Methionin, Lysin, Vitamin C, Eisen und Vitamin B3 und B6 notwendig.
Es werden circa 1,2 Micromol/kg/Tag synthetisiert. Das sind umgerechnet circa 12 mg für einen normalgewichtigen Menschen.
Dieser Wert kann allerdings deutlich schwanken. Denn die Enzym-Aktivität (s. o.) wird durch viele Faktoren reguliert. Besonders wichtig scheint in diesem Zusammenhang der molekulare Schalter PPARalpha zu sein. PPARalpha reguliert, einfach ausgedrückt, den Fettstoffwechsel. Auch in der Leber. Steigt der Fettsäure-Turn-Over, erkennt die Leber ebendas und reguliert via PPARalpha nicht nur die Fettverbrennung, sondern auch die L-Carnitin-Synthese. 1 Das liegt auf der Hand.
Allerdings: Das Leber-PPARalpha wird bei vielen Individuen deutlich unterdrückt sein, denn eine klassische Western Diet (Zucker, Fett und viele Kalorien) überfrachtet die Leber mit Substraten, die wiederum PPARalpha ausknipst. 2 Dies kommt insbesondere bei Leberverfettung zum Tragen. Was dabei das Ei bzw. die Henne war, lässt sich, wie oft, nicht eindeutig sagen.
Ein fehlregulierter Carnitin-Stoffwechsel zeigt sich:
- im Alter
- bei Fettleibigkeit
- bei Sonderformen der Ernährung (z. B. Veganismus)
- bei Leberstoffwechselstörungen (z. B. Fettleber)
- und anderen anomalen Stoffwechselsituationen
(Vgl., z. B. 3)
L-Carnitin hat heute keine gute Reputation, wurde jahrelang in ein falsches Licht gerückt und in eine Rolle gedrückt. L-Carnitin ist jedoch weit mehr als das, was generell angenommen wird.
Bekannt ist, dass L-Carnitin dabei hilft, Fettsäuren über die mitochondriale Membran zu transportieren, sodass diese im Inneren der Mitochondrien, via ß-Oxidation, abgebaut werden können.
Die weniger bekannte Rolle des L-Carnitins ist das Puffern von Acyl- und Acetyl-Gruppen. Was das ist und welche Rolle das spielt, darauf werden wir gleich eingehen.
Der mitochondriale L-Carnitin-Stoffwechsel
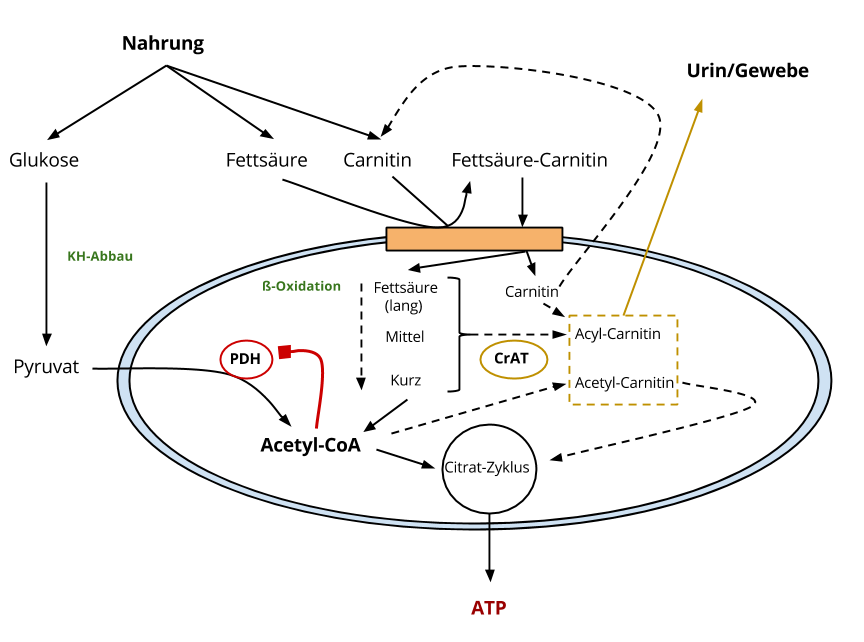
Ich möchte kurz diese Abbildung besprechen und aufzeigen, wie Carnitin im mitochondrialen Stoffwechsel involviert ist.
Die Abbildung zeigt das Zellinnere, genauer: Ein Blick auf das Mitochondrium und das Innere desselben.
L-Carnitin transportiert Fettsäuren ins Mitochondrien-Innere
Auf beiden mitochondrialen Membranen sitzen Proteine, die insgesamt zum gleichen Komplex gehören, ich nenne es der Einfachheit halber „Carnitin-Komplex“ (orange eingezeichnet). Dieser Komplex soll den Transport von Fettsäuren ins Mitochondrien-Innere ermöglichen.
Dieser Enzymkomplex sorgt dafür, dass freie Fettsäuren sich mit L-Carnitin verbinden. Diese Reaktion und somit die entsprechende Fettsäure-Carnitin-Verbindung, ermöglicht den Eintritt ins Mitochondrium. Im Innersten des Mitochondriums werden diese Fettsäuren wieder vom Carnitin getrennt. Carnitin kann nun für weitere Aufgaben im Mitochondrium bereitstehen oder wieder nach außen wandern.
Fettsäuren (ß-Oxidation) und Glukose werden zu Acetyl-CoA abgebaut
Die noch langkettigen Fettsäuren werden via ß-Oxidation zu kürzeren Fettsäuren abgebaut. Fettsäuren nennt man in diesem Zusammenhang „Acyle“. So gibt es logischerweise langkettige Acyle (long-chain acyl), mittelkettige Acyle (medium-chain acyl) und kurzkettige Acyle (short-chain acyl).
Das kürzeste dieser Acyle nennt man Acetyl.
Anmerkung: Richtigerweise hängt noch ein CoA hintendran (z. B. long-chain acyl-CoA), aber an dieser Stelle eine untergeordnete Rolle.
Das Endprodukt der ß-Oxidation, aber auch der Glukose-Oxidation, ist Acetyl-CoA. Dieses Acetyl-CoA kann in den Citrat-Zyklus eingehen und am Ende entsteht ATP. Der Energieträger des Lebens.
Fettsäure-Oxidation hemmt den Kohlenhydratstoffwechsel
Fluten Fettsäuren im Zuge einer Mahlzeit an, so erhöht sich der Flux durch die ß-Oxidation.
Bei Individuen, die Fettsäuren gut oxidieren, entsteht am Ende Acetyl-CoA. Dieses Acetyl-CoA bremst das Master-Enzym des Kohlenhydrat-Stoffwechsels namens Pyruvat-Dehydrogenase (kurz: PDH) aus. PDH wird aufgrund dieser Fettsäure-Fracht also ausgeknipst, weswegen der Kohlenhydrat-Flux deutlich reduziert ist. Das ist gut und wichtig.
Bei Individuen, die Fettsäuren schlecht oxidieren, entsteht am Ende auch Acetyl-CoA, allerdings fallen sehr viele „unvollständig oxidierte Fettsäuren“ an. Das kannst du anhand der Grafik erkennen. Die Maschinerie spuckt alle möglichen Längen von Acylen (= Fettsäuren) aus. Diese reichern sich an und dienen der Synthese von anderen Lipid-Spezies, die letztendlich das Insulin-Signal blockieren und den zellulären Glukose-Eintritt verhindern (nicht eingezeichnet). Dazu gleich mehr.
L-Carnitin und die metabolische Entgleisung
L-Carnitin wirkt als physiologischer Acetyl- und Acyl-Puffer.
L-Carnitin und Acetyl-CoA
Reichert sich extrem viel Acetyl-CoA im Zuge der Fettsäure-Oxidation an, wird die Kohlenhydrat-Oxidation via PDH ausgebremst. 4 5 Dies passiert vor allem dann, wenn zu viel Acetyl-CoA relativ zur Acetyl-CoA-Verarbeitung via Citrat-Zyklus anfällt (= zu viel Substrate umgesetzt wurden relativ zum Verbrauch) und zu wenig CoA regeneriert wird.
L-Carnitin wirkt hier als Puffer und bindet überschüssige Acetyl-Moleküle, regeneriert dabei CoA-Moleküle. 6
(Anmerkung: Acetyl-, sowie Acyl-Moleküle, die wir im Verlauf besprechen, werden durch das Enzym Carnitin-Acetyltransferase bzw. Carnitin-Acyltransferase, kurz CrAT, übertragen.)
Dies hebt die Hemmung der PDH-Enzym-Aktivität auf. Es zeigt sich, dass eine L-Carnitin-Gabe den Pyruvat-Flux sehr deutlich erhöhen kann. (Daraus folgt: Stärkerer nicht-oxidativer Glukose-Verbrauch.) 7
Gleichzeitig könnte L-Carnitin die Acetyl-Gruppen später wieder abgeben und würde somit als kleines Energie-Reservoir dienen. Denkbar wäre beispielsweise eine Donor-Funktion bei der Acetyl-Cholin-Synthese. Darüber hinaus kann L-Carnitin die Acetyl-CoA-Moleküle auch via Pyruvat-Carboxylase als Intermediat des Citrat-Zyklus einschleusen. Tatsächlich ist beschriebene L-Carnitin-Mechanismus genial: Acetyl-Carnitin, ein energiereiches Substrat, kann auch den Ort des Geschehens verlassen und anderen Geweben als Energieträger dienen (z. B. dem Gehirn). Die Studien zu Acetyl-L-Carnitin sind bekannt — denn diese Substanz gibt es als Ergänzungsmittel zu kaufen.
Ursprünglich bekannt wurde diese Pufferfunktion durch die Beobachtung, dass manche Spezies einen hohen L-Carnitin-Gehalt in glykolytischen Muskelfasern aufweisen. Hier dient L-Carnitin in keinster Weise dem Fettsäure-Transport (da keine Fettsäure-Oxidation nötig), sondern dem Acetyl-CoA-Puffer, was verhindert, dass der glykolytische Muskel zu stark übersäuert (Laktat-Bildung). Denn: Unverarbeitetes Pyruvat kann, wenn nicht zu Acetyl-CoA via PDH verstoffwechselt, zu Laktat verarbeitet werden. Die L-Carnitin-Gabe (an Menschen) kann diesen Effekt mimen, wobei die anaerobe Glykolyse unterdrückt und die oxidative Energiegewinnung gefördert wird. 8
Es scheint so zu sein, dass L-Carnitin vor allem Acetyl-CoA-Moleküle des Kohlenhydrat-Stoffwechsels bindet und erklärt somit auch, warum L-Carnitin den Kohlenhydrat-Flux erhöht.9 10
L-Carnitin und Acyl-CoA
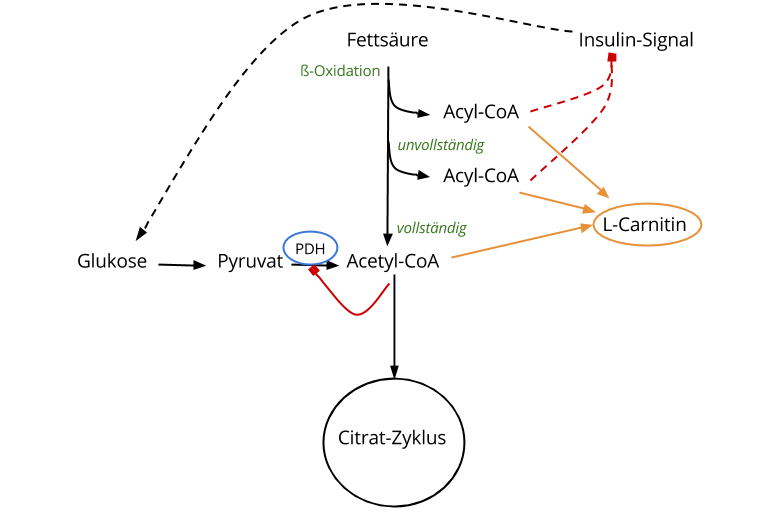
Eine unvollständige ß-Oxidation, wie wir das bei Insulinresistenten beobachten, lässt — wie vorhin besprochen — Acyl-CoA-Moleküle aller Größen entstehen. 11 Diese allerdings dienen der Synthese von anderen Lipid-Spezies, die im Zuge der Insulinresistenz das zelluläre Insulin-Signal blockieren und somit den zellulären Glukose-Eintritt verhindern. 12 13
Es ist bekannt, dass L-Carnitin ebendiese Acyl-CoA-Moleküle binden kann. Heute ist man sich sicher, dass diese Acyl-Carnitin-Verbindung ein protektiver Mechanismus ist, der die Zelle vor Stress (= Anreicherung von Lipid-Spezies) schützt. 14 15
Diese unvollständig oxidierten Fettsäuren verbrauchen das freie Carnitin und binden es. Daraus folgt zwar, dass das Mitochondrium den Zustand teil- und zeitweise puffern kann, aber gleichzeitig raubt es der Zelle das freie Carnitin, das dann nicht mehr zur Verfügung steht.
Da L-Carnitin zunächst durchs Zytosol muss, um ins Mitochondrien-Innere zu gelangen, erfährt das CPT1-System (äußere mitochondriale Membran) eine Carnitin-Präferenz. Das heißt: Der Eintritt von Fettsäuren in die Mitochondrien bleibt immer vorrangig. Sind Fettsäuren aber einmal im Mitochondrium, müssen sie auch verarbeitet werden.
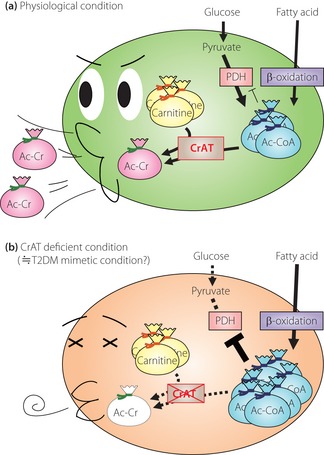
Der metabolische Druck auf das Mitochondrium wächst also proportional zum Fettsäure-Angebot. Die Mitochondrien zeigen eine Überlastung, es fallen vermehrt unvollständig oxidierte Fettsäuren an, die werden — wie beschrieben — an Carnitin gebunden (=> Pufferfunktion), was das freie Carnitin in den Mitochondrien raubt.
Das Bemerkenswerte ist, dass ein hoher Fettsäure-Flux (z. B. bei Fettleibigkeit und einer fettreichen Ernährung) das Enzym (Carnitin-Acyltransferase, CrAT) hemmt, das diese Acyle auf Carnitin überträgt. Insbesondere der Palmitinsäure-Abkömmling Palmitoyl-CoA scheint hier eine große Rolle zu spielen.
Mit der Zeit reichern sich diese Fettsäure-Abkömmlinge an, hemmen die Carnitin-Acyltransferase-Aktivität und setzen somit die Puffer-Funktion des Carnitins außer Kraft. Die Enzym-Aktivität wird zudem negativ reguliert durch die Tatsache, dass die Carnitin-Konzentration in den Mitochondrien fällt.
Denn: Carnitin selbst reguliert die Gen-Expression der CrAT — soll heißen, dass eine ausreichende Carnitin-Konzentration gegeben sein muss, damit CrAT ausreichend gebildet wird. 16
Daraus folgt das paradoxe Bild, dass der Fettsäure-Eintritt in die Mitochondrien ungebremst bleibt, aber die mitochondriale Kapazität bezüglich des optimalen Umgangs mit Fettsäuren dramatisch fällt.
In anderen Worten: Es ist der Abfall der mitochondrialen L-Carnitin-Konzentration, das eine eingeschränkte metabolische Flexibilität als Folge hat! (Vgl. 17)
Bestätigt werden diese Annahmen durch ein Mäuse-Modell, bei dem die muskelspezifische CrAT (Carnitin-Acyltransferase) fehlt: Es zeigt sich eine dramatisch eingeschränkte metabolische Flexibilität, vor allem beim Switch vom Fett- auf den Kohlenhydratstoffwechsel. Z. B. nach einer kohlenhydratreichen Mahlzeit. 18
Dies deckt sich mit Versuchen am Menschen und an Tieren, bei denen eine L-Carnitin-Supplementation den Pyruvat-Flux (= den Kohlenhydrat-Stoffwechsel verbessert; Marker für die metabolische Flexibilität) und die Acyl-Carnitin-Konzentration (als Marker für die Funktion der Carnitin-Acyltransferase) deutlich erhöht. 19
Auch eine Überexpression der CrAT in menschlichen Muskelzellen zeigt ein deutlich besseres Glukose-Handling und ein reziprokes Normalisieren der zu intensiven Fettsäure-Oxidation. 20
Bei fettleibigen Ratten ist die metabolische Flexibilität nicht gegeben und es zeigen sich niedrige L-Carnitin-Zellwerte. Eine L-Carnitin-Gabe stellt die metabolische Flexibilität wieder her, kehrt diese Anomalie also um. 21
Weitere Resultate
In beiden Fällen, sowohl bei der Acyl-CoA- als auch bei de Acetyl-CoA-Pufferung, wird das CoA-Molekül frei, das extrem wichtig ist, um aus Pyruvat (Endprodukt des KH-Stoffwechsels), Acetyl-CoA zu generieren. Gleichzeitig wird CoA im Citrat-Zyklus benötigt. Ein CoA-Mangel also, würde nicht nur den Kohlenhydrat-Stoffwechsel, sondern auch den Citrat-Zyklus ausbremsen, was das zelluläre Chaos verstärkt.
L-Carnitin als Gen-Regulator
Vor einigen Jahren konnte anhand menschlicher Muskeln, bei einem In-Vivo-Versuch, gezeigt werden, dass die L-Carnitin-Gabe mehr als 70 Gene des Zellstoffwechsels reguliert. Darunter auch Enzyme des Citrat-Zyklus, der ß-Oxidation, des Fettstoffwechsels und Gene, die die mitochondriale Gesundheit regulieren. 22
(Diese Arbeit legte nahe, dass Carnitin wohl rate-limiting wirkt bezüglich des Fettsäure-Transportes und zeigte eine dramatische Erhöhung des Fettsäure-Flux [vierfach] über die mitochondriale Membran. Und das trotz der Tatsache, dass die CPT1-Enzym-Aktivität nicht zunahm.)
Eine andere Arbeit zeigt, dass eine L-Carnitin-Gabe den mit der Insulinresistenz einhergehenden Muskelfaser-Switch (oxidativ -> glykolytisch) hemmt und die oxidative Kapazität somit aufrecht erhalten kann. 23
Wie oben bereits angeschnitten, scheint die L-Carnitin-Verfügbarkeit maßgeblich die im Carnitin-Stoffwechsel involvierten Proteine zu regulieren, darunter CPT1 (äußere mitochondriale Membran) und das hier ausführlichst besprochene CrAT. Das passt zum Bild der hier geschilderten Sachverhalte.
Andernorts wird nahe gelegt, dass L-Carnitin maßgeblich die Funktion und Morphologie des braunen Fettgewebes reguliert. 24 Auch hieran wird deutlich, wie ein Konzentrationsabfall des freien Carnitins zu einer systemischen metabolischen Dysfunktion führen könnte.
Kurzum: L-Carnitin ist weit mehr als ein Fettsäure-Transporter. L-Carnitin selbst (oder die daraus gebildeten Substanzen) ist Regulator des zellulären Stoffwechsels und scheint ein überragendes Beispiel dafür zu sein, wie die Umwelt (hier: Carnitin-Verfügbarkeit) Einfluss auf das Genom nimmt. Daher ist die Frage berechtigt, ob und inwieweit eine deutliche Reduktion des freien Carnitins für eine Verschlechterung des Zell-Milieus sorgt.
Abschließende Worte und Aussicht
Diese hier vorgestellte Theorie ist in meinen Augen bahnbrechend. Bisher wurden immer nur gewisse Phänomene angesprochen. Heute wird erstmals auch ein gemeinsamer Nenner genannt: Carnitin-Acyltransferase.
Die Funktion der Carnitin-Acyltransferase ist bei Insulinresistenten eingeschränkt, weswegen die Pufferfunktion von L-Carnitin entfällt — gleichwohl scheint L-Carnitin dieser Entgleisung entgegen zu wirken und kann die Wirkung wiederherstellen.
Es zeigt sich, dass L-Carnitin eine Schlüsselsubstanz im mitochondrialen Energiestoffwechsel ist.
Sollten sich die Ergebnisse bewahrheiten und in vivo am Menschen nachweisen, hätten wir hier ein sehr mächtiges therapeutisches Tool. In der Tat wäre dies kein Tool mehr, sondern die Korrektur einer fehlgeleiteten Situation.
Es wäre sicher spannend zu erfahren, ob es genetische Unterschiede bezüglich der Regulation des CrAT-Gens und/oder ob es unterschiedliche Kapazitäten bezüglich des (Acyl-)Carnitin-Stoffwechsels gibt, die Unterschiede zwischen einzelnen Individuen erklären könnten. Auch hier gibt es bereits Evidenz: Ratten, die von Haus aus eine hohe Ausdauerleistungsfähigkeit (high capacity) zeigen, sind — im Gegensatz zu Ratten mit niedriger Ausdauerleistungsfähigkeit (low capacity) — geschützt vor Glukose-Intoleranz (durch ein High-Fat-Feeding). Der Grund ist simpel: Bei High-Capacity-Ratten fällt das freie Carnitin nicht ab! 25
Dies legt bereits nahe, dass Individuen wohl unterschiedliche Bedürfnisse haben und insbesondere diejenigen anfällig sind für metabolische Entgleisungen, die — vielleicht genetisch — beeinträchtigt sind bezüglich ihrer Ausdauerleistungsfähigkeit (diese sei an dieser Stelle nicht genauer definiert).
Literatur
- Makowski, L.; Noland, R. C.; Koves, T. R. u. a. (2008): „Metabolic profiling of PPAR -/- mice reveals defects in carnitine and amino acid homeostasis that are partially reversed by oral carnitine supplementation“. In: The FASEB Journal. 23 (2), S. 586-604, DOI: 10.1096/fj.08-119420.
- Nagai, Yoshio; Nishio, Yoshihiko; Nakamura, Takaaki u. a. (2002): „Amelioration of high fructose-induced metabolic derangements by activation of PPARα“. In: American Journal of Physiology – Endocrinology And Metabolism. 282 (5), S. E1180-E1190, DOI: 10.1152/ajpendo.00471.2001.
- Flanagan, Judith L; Simmons, Peter A; Vehige, Joseph u. a. (2010): „Role of carnitine in disease“. In: Nutrition & Metabolism. 7 (1), S. 30, DOI: 10.1186/1743-7075-7-30.
- BOWKER-KINLEY, Melissa M.; DAVIS, I. Wilhelmina; WU, Pengfei u. a. (1998): „Evidence for existence of tissue-specific regulation of the mammalian pyruvate dehydrogenase complex“. In:Biochem. J.. 329 (1), S. 191-196, DOI: 10.1042/bj3290191.
- Hue, L.; Taegtmeyer, H. (2009): „The Randle cycle revisited: a new head for an old hat“. In: AJP: Endocrinology and Metabolism. 297 (3), S. E578-E591, DOI: 10.1152/ajpendo.00093.2009.
- Miyata, Yugo; Shimomura, Iichiro (2013): „Metabolic flexibility and carnitine flux: The role of carnitine acyltransferase in glucose homeostasis“. In: Journal of Diabetes Investigation. 4 (3), S. 247-249, DOI: 10.1111/jdi.12064.
- Uziel, Graziella; Garavaglia, Barbara; Di Donato, Stefano (1988): „Carnitine stimulation of pyruvate dehydrogenase complex (PDHC) in isolated human skeletal muscle mitochondria“. In: Muscle & Nerve. 11 (7), S. 720-724, DOI: 10.1002/mus.880110708.
- Wall, Benjamin T.; Stephens, Francis B.; Constantin-Teodosiu, Dumitru u. a. (2011): „Chronic oral ingestion of l -carnitine and carbohydrate increases muscle carnitine content and alters muscle fuel metabolism during exercise in humans“. In: The Journal of Physiology. 589 (4), S. 963-973, DOI: 10.1113/jphysiol.2010.201343.
- Abdel-aleem, S (1996): „Regulation of Fatty Acid Oxidation by Acetyl-CoA Generated from Glucose Utilization in Isolated Myocytes“. In: Journal of Molecular and Cellular Cardiology. 28 (5), S. 825-833, DOI: 10.1006/jmcc.1996.0077.
- Abdel-aleem, S (1995): „Stimulation of Non-oxidative Glucose Utilization by?-carnitine in Isolated Myocytes“. In: Journal of Molecular and Cellular Cardiology. 27 (11), S. 2465-2472, DOI: 10.1006/jmcc.1995.0234.
- Koves, Timothy R.; Ussher, John R.; Noland, Robert C. u. a. (2008): „Mitochondrial Overload and Incomplete Fatty Acid Oxidation Contribute to Skeletal Muscle Insulin Resistance“. In: Cell Metabolism. 7 (1), S. 45-56, DOI: 10.1016/j.cmet.2007.10.013.
- COONEY, G. J.; THOMPSON, A. L.; FURLER, S. M. u. a. (2002): „Muscle Long-Chain Acyl CoA Esters and Insulin Resistance“. In: Annals of the New York Academy of Sciences. 967 (1), S. 196-207, DOI: 10.1111/j.1749-6632.2002.tb04276.x.
- Zhang, D.; Liu, Z.-X.; Choi, C. S. u. a. (2007): „Mitochondrial dysfunction due to long-chain Acyl-CoA dehydrogenase deficiency causes hepatic steatosis and hepatic insulin resistance“. In:Proceedings of the National Academy of Sciences. 104 (43), S. 17075-17080, DOI: 10.1073/pnas.0707060104.
- Koves, Timothy R.; Ussher, John R.; Noland, Robert C. u. a. (2008): „Mitochondrial Overload and Incomplete Fatty Acid Oxidation Contribute to Skeletal Muscle Insulin Resistance“. In: Cell Metabolism. 7 (1), S. 45-56, DOI: 10.1016/j.cmet.2007.10.013.
- Miyata, Yugo; Shimomura, Iichiro (2013): „Metabolic flexibility and carnitine flux: The role of carnitine acyltransferase in glucose homeostasis“. In: Journal of Diabetes Investigation. 4 (3), S. 247-249, DOI: 10.1111/jdi.12064.
- Godárová, Alzbeta; Litzlbauer, Elke; Brunner, Sylvia u. a. (2005): „L-Carnitine Regulates mRNA Expression Levels of the Carnitine Acyltransferases – CPT1A, CPT2, and CRAT“. In: Monatshefte für Chemie – Chemical Monthly. 136 (8), S. 1349-1363, DOI: 10.1007/s00706-005-0336-5.
- Seiler, S. E.; Martin, O. J.; Noland, R. C. u. a. (2014): „Obesity and lipid stress inhibit carnitine acetyltransferase activity“. In: The Journal of Lipid Research. 55 (4), S. 635-644, DOI: 10.1194/jlr.m043448.
- Muoio, Deborah M.; Noland, Robert C.; Kovalik, Jean-Paul u. a. (2012): „Muscle-Specific Deletion of Carnitine Acetyltransferase Compromises Glucose Tolerance and Metabolic Flexibility“. In: Cell Metabolism. 15 (5), S. 764-777, DOI: 10.1016/j.cmet.2012.04.005.
- Seiler, S. E.; Martin, O. J.; Noland, R. C. u. a. (2014): „Obesity and lipid stress inhibit carnitine acetyltransferase activity“. In: The Journal of Lipid Research. 55 (4), S. 635-644, DOI: 10.1194/jlr.m043448.
- Noland, R. C.; Koves, T. R.; Seiler, S. E. u. a. (2009): „Carnitine Insufficiency Caused by Aging and Overnutrition Compromises Mitochondrial Performance and Metabolic Control“. In: Journal of Biological Chemistry. 284 (34), S. 22840-22852, DOI: 10.1074/jbc.m109.032888.
- Noland, R. C.; Koves, T. R.; Seiler, S. E. u. a. (2009): „Carnitine Insufficiency Caused by Aging and Overnutrition Compromises Mitochondrial Performance and Metabolic Control“. In: Journal of Biological Chemistry. 284 (34), S. 22840-22852, DOI: 10.1074/jbc.m109.032888.
- Stephens, Francis B.; Wall, Benjamin T.; Marimuthu, Kanagaraj u. a. (2013): „Skeletal muscle carnitine loading increases energy expenditure, modulates fuel metabolism gene networks and prevents body fat accumulation in humans“. In: The Journal of Physiology. 591 (18), S. 4655-4666, DOI: 10.1113/jphysiol.2013.255364.
- Couturier, Aline; Ringseis, Robert; Mooren, Frank-Christoph u. a. (2014): „Correction: Carnitine supplementation to obese Zucker rats prevents obesity-induced type I to type II muscle fiber transition and favors an oxidative phenotype of skeletal muscle“. In: Nutrition & Metabolism. 11 (1), S. 16, DOI: 10.1186/1743-7075-11-16.
- Ozaki, Kiyokazu; Sano, Tomoya; Tsuji, Naho u. a. (2011): „Carnitine is necessary to maintain the phenotype and function of brown adipose tissue“. In: Lab Invest. 91 (5), S. 704-710, DOI: 10.1038/labinvest.2011.6.
- Noland, R. C.; Koves, T. R.; Seiler, S. E. u. a. (2009): „Carnitine Insufficiency Caused by Aging and Overnutrition Compromises Mitochondrial Performance and Metabolic Control“. In: Journal of Biological Chemistry. 284 (34), S. 22840-22852, DOI: 10.1074/jbc.m109.032888.
8 comments On Carnitin: So reguliert es deinen Stoffwechsel
Carnitin ist super keine Frage, aber die Nebenwirkung ist ekelhafter, nach Fisch und Pipi stinkender Körpergeruch. Dieser Geruch geht sogar in die Kleidung über und in die Bettwäsche. Dort hält er sich tagelang.
Hi,
Vielen Dank für den Artikel.
Leider verstehe ich nicht, was genau nun passieren soll und zu welchem Zweck?
Ich nehme L-Carnitin vor dem Sport und habe gefühlt mehr Leistung. Ohne Sport merke ich nichts. Will Abnehmen, das ist mein Ziel.
Falsche Annahme?
Hi Chris,
was ist mit der Aussage, das oral verabreichtes Carnitin als NEM auf Grund enzymatischer Inhibition gar nicht in der Zelle ankommt sondern nur in natura, d.h. mit Fleischverzehr wirkt, da dort das abbauende Enzym gehemmt wird.
Hi Uwe,
du meinst Carnosin, nicht Carnitin.
LG, Chris
Hallo Chris, wenn das wirklich eine „bahnbrechende“ Hypothese ist, wie Du es nennst, dann diskutiere sie auch mal mit Leuten Deines Faches. Hier tummeln sich ja im wesentlichen Hobby-Forscher. Ich schätze jedenfalls, dass nicht allzu viele Deiner Leser Biochemie und Medizin studiert haben.
Vielleicht kannst Du im Artikel noch darauf eingehen, wie die Fettsäuren in der Zelle das Insulinsignal blockieren. Du schreibst hierzu ja nur : „Diese allerdings dienen der Synthese von anderen Lipid-Spezies, die im
Zuge der Insulinresistenz das zelluläre Insulin-Signal blockieren und
somit den zellulären Glukose-Eintritt verhindern“.
Ist bekannt, wie das genau funktioniert?
Besten Gruß
Peter
Hi Peter,
ich weiß jetzt nicht so genau, was du mir mit dem Beitrag sagen willst.
Soll ich daraus schlussfolgern, dass ich nichts mehr in dieser Richtung schreiben soll, weil die Leute nichts verstehen?
Ist ein bisschen eine paradoxe Forderung in Anbetracht der Tatsache, dass es ja mein Blog ist und ich hier seit Beginn meine Gedanken niederschreibe.
Und … dass ich es hier poste, schließt ja keinesfalls aus, dass ich es nicht auch mit Kollegen diskutiere?!
Zum Acyl-CoA, Lipid-Spezies und Insulin-Signal: Acyl-CoA dient der Synthese von DAG (Diaglycerine), was eine Substanz namens PKC aktiviert und die „falsche“ Aminosäure am Insulin-Rezeptor-Substrat phosphoryliert und somit das Insulin-Signal blockiert.
(Kann hier im Blog übrigens auch nachlesen.)
LG, Chris
Hallo Chris, nein, dass sollte keine Aufforderung zur Senkung des
Niveaus sein. War nur so ein Gedanke. Vielleicht überflüssig. Wenn ja,
dann umso besser :).
Zur IR: Es hat mich gewundert, weil sich
Dein Post so liest, als ob der nachfolgende Mechanismus, der zur IR
führt, trivial wäre. Oft liest man, dass die Mechanismen, die zu Typ-2
Diabetes führen noch schlecht verstanden sind.
Grüße, Peter
Hi Peter,
okay, okay! :-)
Trivial ist es nicht. Biochemisch komplex sicher. Aber die Mechanismen sind sehr gut studiert und es ergibt sich ein sehr deutliches Bild.
Natürlich ist es noch einmal komplexer, wenn wir Entzündungen etc. in die Gleichung miteinbeziehen — die spielen sicher auch eine große Rolle über einen anderen Mechanismus.
Aber die Lipid-induzierte IR ist sehr gut studiert.
Liebe Grüße,
Chris